


Effects of Growth Environment on Bacterial Community Diversity of Procambarus clarkia
-
摘要: 为探索生长环境对克氏原螯虾(Procambarus clarkii)细菌群落多样性的影响,本实验利用Illumina MiSeq测序技术,对克氏原螯虾尾肉、养殖水体及周围土壤进行基因序列测序,分析小龙虾虾肉与其生长环境之间的相关性。多样性分析结果表明,土壤的微生物多样性高于小龙虾和水体多样性,分析三组样品在不同分类水平的相对丰度发现,小龙虾、土壤和水体中有相对固定的优势菌群,在门类为变形菌门(Proteobacteria)和拟杆菌门(Bacteroidetes),α-变形菌纲(Alphaproteobacteria)、β-变形菌纲(Betaproteobacteria)和δ-变形菌纲(Deltaproteobacteria)等在小龙虾尾肉、土壤及水体中的平均相对丰度较高,而在科和属水平上无共有优势菌群。PICRUSt2代谢功能预测结果表明,3组样品在有氧呼吸(aerobic respiration I)、脂肪酸合成(fatty acid elongation)、氨基酸生物合成(amino acid synthesis)和核糖核苷酸代谢途径(nucleotide sugar metabolism)中代谢通路丰度最高。研究结果进一步解释了生长环境和克氏原螯虾微生物群落结构间的相关性和差异性。Abstract: In order to explore the effects of growth environment on the bacterial community diversity of Procambarus clarkia, we used Illumina miseq sequencing technology to sequence the tail meat of Procambarus clarkii, the culture water and the soil around the culture environment, and analyzed the correlation between the meat and the growth environment of Procambarus clarkii. The results showed that the diversity of Proteobacteria, Bacteria and Proteobacteria were higher than that of Proteobacteria and Bacteria in water. The average relative abundance of Deta Proteobacteria and Delta Proteobacteria were higher in crayfish tail meat, soil and water, but there was no common dominant flora at the family and genus level. The results of PICRUSt2 metabolic function prediction showed that the metabolic pathway abundance of three groups of samples was the highest in aerobic respiration (I), fatty acid synthesis (fat acid synthesis), amino acid biosynthesis (amino acid synthesis) and nucleoside sugar metabolism pathway (nucleotide sugar metabolism). The results of the study further explained the correlation and difference between the growth environment and the microbial community structure of Procambarus clarkia.
-
Keywords:
- Procambarus clarkii /
- soil /
- water /
- flora structure /
- high-throughput sequencing
-
小龙虾学名克氏原螯虾(Procambarus clarkii),是中国水产养殖中主要物种之一。近年来,小龙虾养殖面积和养殖产量持续增长。根据2020年小龙虾产业发展报告,2019年我国小龙虾养殖总产量达208.96万吨,与2018年相比,同比增长27.52%[1]。小龙虾-稻田循环系统是小龙虾主要养殖方式[2],在此生态系统中,生产力、养分循环及水质离不开环境微生物的作用[3]。小龙虾养殖系统中细菌群落组成对小龙虾体内细菌微生物区系有很大影响,体内微生物区系在动物的营养、免疫及抗病等方面发挥重要作用[4]。池塘底泥中含有丰富的营养物质,在水生生物的生长过程中承担着营养物质的再循环作用,因此菌落结构复杂。秦伟等[5]发现在池塘底泥中变形菌门是克氏原螯虾的主要优势菌群,随着养殖时间的变化,底泥中菌群的丰富度及多样性逐渐降低,而长毛对虾的养殖水体中细菌优势种群为蓝细菌,芽孢杆菌在目分类水平上占有较高比例[6]。研究发现,小龙虾携带的沙门氏菌、金黄色葡萄球菌及副溶血性弧菌等致病菌与其生活的水体环境密切相关[7-8]。
基因组测序和高通量测序(High-through putsequencing,HST)技术在描述多样性环境中的微生物群落方面取得了突破性进展[9-10]。基因组学克服了传统微生物分离鉴定技术,采用直接提取DNA的方法检测微生物菌落的多样性,利用基因组序列分析对比结果,对微生物进行生物信息学预测[11]。Illumina MiSeq测序仪已被用于16S rRNA序列研究,能够更好地检测和分析自然界中的次优势菌和稀有细菌分类群[12-13]。16S rRNA高通量测序分析表明[14],变形杆菌、类杆菌和梭杆菌是克氏原螯虾肠道微生物区系中的优势菌群,其中变形杆菌参与土壤中碳、氮的循环。汤纯等[15]运用Illumina Miseq宏基因组探究了小龙虾不同部位的菌相组成发现,在小龙虾尾肉中主要微生物为希瓦氏菌属、拟杆菌属,并发现水体环境可能对小龙虾携带的菌群多样性影响较高。
目前关于小龙虾微生物的研究主要集中于养殖水体中的微生物多样性及小龙虾肠道微生物菌落结构研究,关于生长水体及土壤对小龙虾微生物群落的影响研究较少。本文以小龙虾生长环境周围的土壤及水体为研究对象,采用Illumina MiSeq高通量测序技术分析生长环境中水体、土壤及小龙虾细菌群落组成,探讨养殖水体及周围土壤中微生物群落与小龙虾细菌群落结构多样性的关系,为改善小龙虾养殖环境,提高小龙虾的食用安全性提供理论参考。
1. 材料与方法
1.1 材料与仪器
活克氏原螯虾(Procambarus clarkii)(体长8~9 cm,体重30±5 g) 2018年7月、8月、9月采集于武汉农科院养殖基地;土壤 2018年采集于武汉农科院养殖基地;水样 2018年采集于武汉农科院养殖基地;微孔滤膜直径47 mm Millipore,USA;DNA分离试剂盒QIAGENSrl Milan,Italy。
SPX-150B-Z型恒温培养箱 上海博迅实业有限公司医疗设备厂;TP600梯度PCR仪 日本Takara Bio Inc;TGL-18M台式冷冻离心机 上海卢湘仪离心机仪器有限公司。
1.2 实验方法
1.2.1 样品采集和DNA的提取
小龙虾在养殖基地采集后30 min内用冰袋运至实验室进行处理,将虾分为三组(C1、C2、C3),尾肉使用液氮速冻后于-80 ℃下冷藏用于DNA提取;小龙虾养殖基地的土壤采集参考伍一宁[16]的方法,利用五点取样法进行土壤采样,去除土壤表面的植被及掉落物,取深度0~10 cm的土壤,过10目筛后混合均匀,分为五组(S1、S2、S3、S4、S5),保存于−80 ℃冰箱用于DNA提取;水样使用灭菌处理后的塑料桶采集,每组样品独立采集三份后混合均匀[17],于实验室经0.22 μm微孔滤膜过滤后,将滤膜转移至无菌离心管中,分为五组(S1、S2、S3、S4、S5),于−80 ℃保存。按照Power Food微生物DNA分离试剂盒(QIAGENSrl,Milan,Italy)的方法进行样品总DNA提取,将其保存于−80 ℃冰箱中。提取的基因组委托北京博云华康基因科技有限公司进行样品检测。
1.2.2 序列质控及生物信息学分析
检测合格的样品构建文库,合格的文库进行cluster制备,利用Illumina平台(Hiseq或Miseq)进行Paired-end测序,用下机得到的数据进行相应的生物信息分析。对数据过滤,获得高质量的Cleandata,通过reads之间的Overlap关系将reads拼接成Tags。序列拼接使用软件FLASH(Fast Length Adjustment of Shortreads,v1.2.11),利用重叠关系将双末端测序得到的reads组装成一条序列,拼接条件设置为:最长匹配长度15 bp,重叠区域允许错配率为0.1,得到高变区的Tags。利用软件USEARCH(v7.0.1090)将拼接好的Tags在97%相似度下将其聚类为用于物种分类的可操作分类单元(Operational Taxonomic Units, OTU)。利用UCHIME(v4.2.40)[18]将PCR扩增产生的嵌合体从OTU代表序列中去除;使用usearch_global方法将所有Tags比对回OTU代表序列,得到每个样品在每个OTU的丰度统计表[19]。根据每个样品的OTU丰度文件,计算每个样品或组别具有的OTU(不考虑OTU丰度,只考虑OTU有无),通过Venn图,展示样品间或组间共有及特有OTU情况。根据每个样品OTU在每个样品的丰度文件计算出每个OTU在每个样品的相对丰度,利用这个丰度信息进行OTU的PCA分析;通过与数据库进行比对,对OTU进行物种分类并分别在门、纲、目、科、属、几个分类等级对各个样品作物种Profiling面积图和柱状图,从纲水平开始,将物种丰度在所有样品均低于0.5%的物种全部合并成Others。最后使用Mothur(v1.31.2)软件计算样品的Alpha多样性值,包括Observed species指数、Chao指数、ACE指数,Shannon指数以及Simpson指数。前4个指数越大,最后一个指数越小,说明样品中的物种越丰富。
1.3 数据处理
使用软件Metastats(http://metastats.cbcb.umd.edu/)(默认)或者R软件(秩和检验,Fisher’s精确检验,卡方检验,t检验,方差检验)进行组间显著性差异分析。
2. 结果与分析
2.1 样品测数据统计及丰度
利用测序获得不同样品的原始序列数据后,对序列进行拼接优化,每组样品中测序结果统计如下(表1),小龙虾、土壤、水体的有效序列分别为77858~95353、47921~74143、61734~90037条,进而对样品序列进行聚类分析,在97%相似的水平下进行OTU生物信息统计,三组样品中的OTUs总数分别为2548~2635个、3274~3737个、1789~2648个。小龙虾中OTUs数量较土壤中OTUs数量显著(P<0.05)减少,说明小龙虾细菌多样性低于土壤细菌多样性。
表 1 测序结果及质量分析Table 1. Sequencing results and quality analysis样品名称 有序数列数量 OTU数量 C1 77858 2619 C2 94552 2548 C3 95353 2635 S1 74143 3497 S2 52230 3734 S3 47921 3737 S4 56102 3278 S5 71336 3274 W1 61734 2596 W2 87664 1789 W3 90037 1871 W4 86817 2648 W5 76027 1813 注:C:小龙虾;S:土壤;W:水体;表2同。 2.2 多样性指数分析
通过单样本多样性(Alpha)分析反映微生物群落的丰度和多样性。样品中群落的丰富度可由Observed species指数、Chao指数和ACE指数来反映[20],简单指群落中物种的数量,而不考虑群落中每个物种的丰度情况;Shannon指数以及Simpson指数反映群落的多样性(Species diversity),受样品群落中物种丰富度(Species richness)和物种均匀度(Species evenness)的影响。相同物种丰富度的情况下,群落中各物种具有越大的均匀度,则认为群落具有越大的多样性。Chao分析表明,小龙虾中细菌群落丰富度范围为2952~3109个,土壤中为3813~4608个,水中为2596~3922个(表2)。利用Shannon指数估算的细菌群落多样性在小龙虾中为5.565~5.760,在土壤中为6.008~6.767,在水中为4.364~5.206(表2)。养殖水体中的Shannon指数及Simpson指数均小于小龙虾及土壤,说明土壤和小龙虾中的微生物群落多样性较水体更为丰富。
表 2 各组样品Alpha多样性统计结果Table 2. Alpha diversity statistical results for each group样品名称 Sobs Chao ACE Shannon Simpson C1 2619 3046.880 3036.437 5.565 0.024 C2 2548 2952.909 2892.741 5.760 0.017 C3 2635 3109.000 3029.552 5.580 0.017 S1 3497 4435.584 4532.497 6.008 0.011 S2 3734 4600.083 4746.686 6.721 0.004 S3 3737 4608.254 4732.640 6.767 0.003 S4 3278 3813.233 3918.379 6.580 0.005 S5 3274 4124.446 4136.989 6.016 0.011 W1 2596 3552.264 3625.095 5.206 0.022 W2 1789 2596.072 3126.695 4.364 0.043 W3 1871 2739.095 3352.675 4.748 0.022 W4 2648 3922.967 4820.739 4.712 0.039 W5 1813 2666.415 2788.272 4.601 0.026 2.3 不同分类水平上的物种注释及分析
2.3.1 基于门分类水平上的物种注释及分析
为获得每个OTU对应物种分类信息,对小龙虾生长环境周围的水体、土壤和虾肉样本的OTU序列进行分类汇总,对应至门、纲、目、科和属5个水平。在所有样品中共检出微生物61个门类,分类至53纲、85目、83科以及74属。
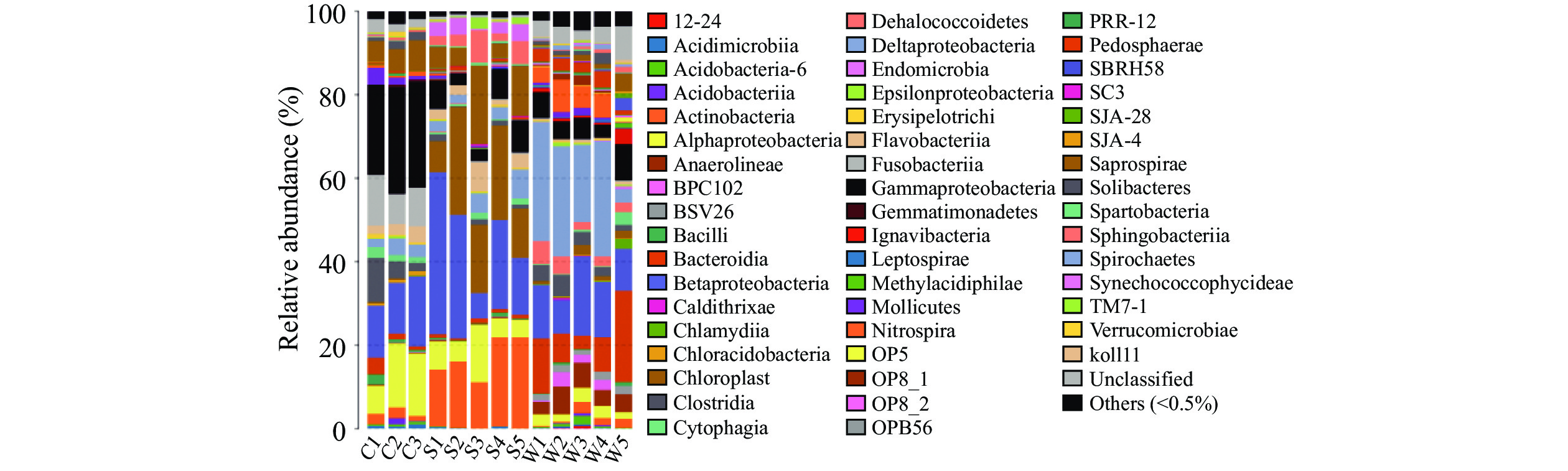
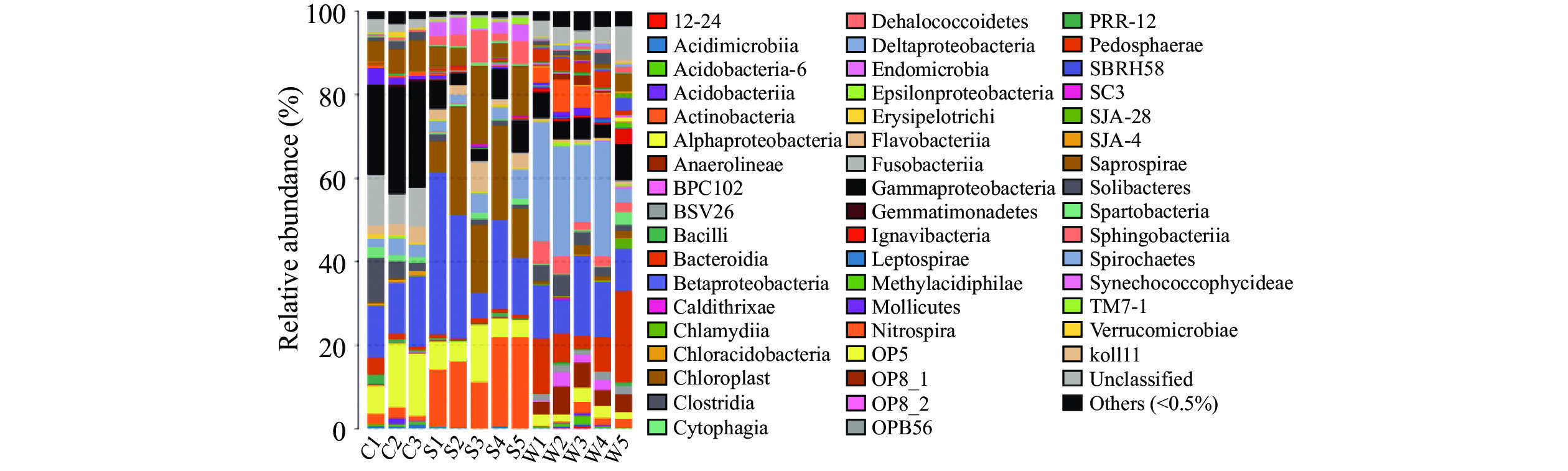
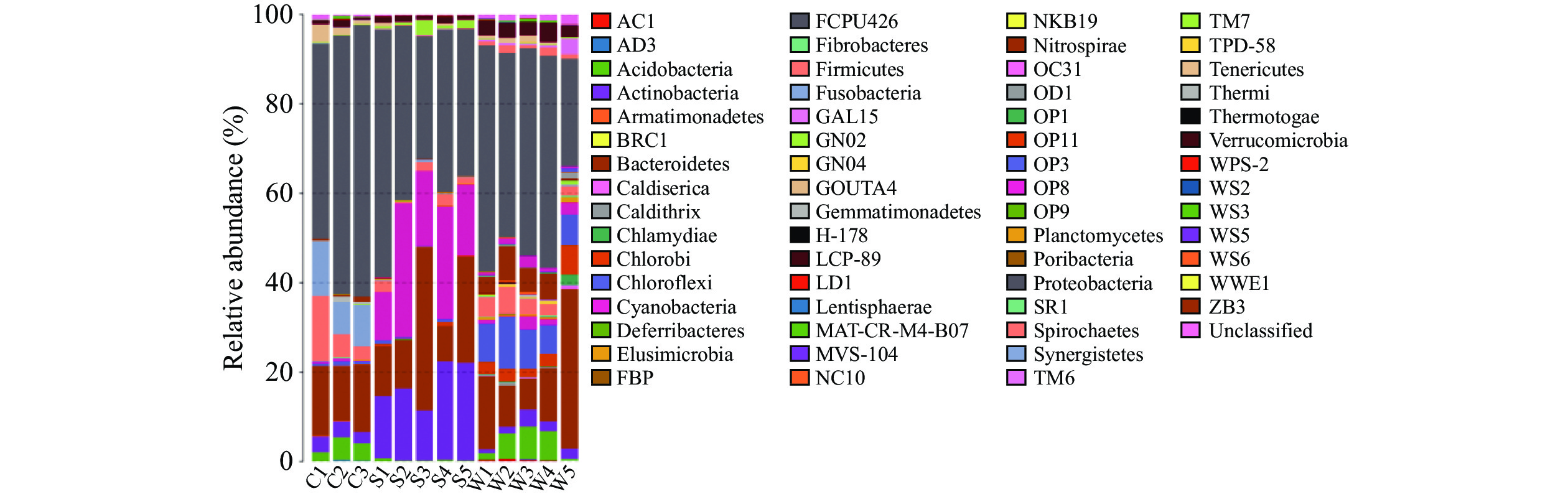
小龙虾尾肉、土壤及水体微生物在门分类水平上的结构分布具有一定规律性:三者的优势门类为变形菌门(Proteobacteria)和拟杆菌门(Bacteroidetes),其中变形菌门占有绝对优势,在小龙虾、土壤及水体中的平均相对丰度依次是54.02%、38.25%、41.94%。不同样品类型在门分类水平上呈现出差异性,小龙虾中梭杆菌门(Fusobacteria)和厚壁菌门(Firmicutes)相对丰度显著(P<0.05)高于土壤和水体;土壤中放线菌门(Actinobacteria)及蓝细菌门(Cyanobacteria)相对丰度高于小龙虾肉和水体,且差异显著(P<0.05);绿弯菌门(Chloroflexi)在虾肉和土壤中相对丰度均小于1%,显著低于水体(P<0.05);酸杆菌门(Acidobacteria)和硝化螺旋菌门(Nitrospirae)在水样中相对丰度亦显著高于底泥(P<0.05)(图1),以上结果说明小龙虾虾肉的菌落结构与养殖环境有密切联系,而土壤中的优势细菌蓝细菌门及水体中优势细菌绿弯菌门在小龙虾中占比较小,说明尽管小龙虾中的菌落结构受到生长环境的影响,但同时也有一定保守性。
![]() 图 1 基于门水平的微生物组成分析柱状图Figure 1. Histogram of microbial composition analysis based on phylum level
图 1 基于门水平的微生物组成分析柱状图Figure 1. Histogram of microbial composition analysis based on phylum level2.3.2 基于纲水平上的物种注释及分析
对微生物在纲分类水平上统计发现,α-变形菌纲(Alphaproteobacteria)、β-变形菌纲(Betaproteobacteria)和δ-变形菌纲(Deltaproteobacteria)等在小龙虾尾肉、土壤及水体中平均相对丰度较高。在小龙虾中,前三大优势纲为γ-变形菌纲(Gammaproteobacteria)、β-变形菌纲(Betaproteobacteria)、α-变形菌纲(Alphaproteobacteria),土壤中为β-变形菌纲(Betaproteobacteria)、放线菌纲(Actinobacteria)及Saprospirae;而水体中δ-变形菌纲(Deltaproteobacteria)、β-变形菌纲(Betaproteobacteria)以及γ-变形菌纲(Gammaproteobacteria)相对丰度较高。微生物纲类在小龙虾尾肉、土壤和水体中的相对丰度存在一定差异。小龙虾尾肉α-变形菌纲(Alphaproteobacteria)及γ-变形菌纲(Gammaproteobacteria)的相对丰度显著高于土壤及水体,而水体中的拟杆菌纲(Bacteroidia)及硝化螺旋菌纲(Nitrospira)则显著较高(图2)。
![]() 图 2 基于纲水平的微生物组成分析柱状图Figure 2. Microbial composition analysis histogram based on class level
图 2 基于纲水平的微生物组成分析柱状图Figure 2. Microbial composition analysis histogram based on class level2.3.3 基于门、科、目水平上的物种注释及分析
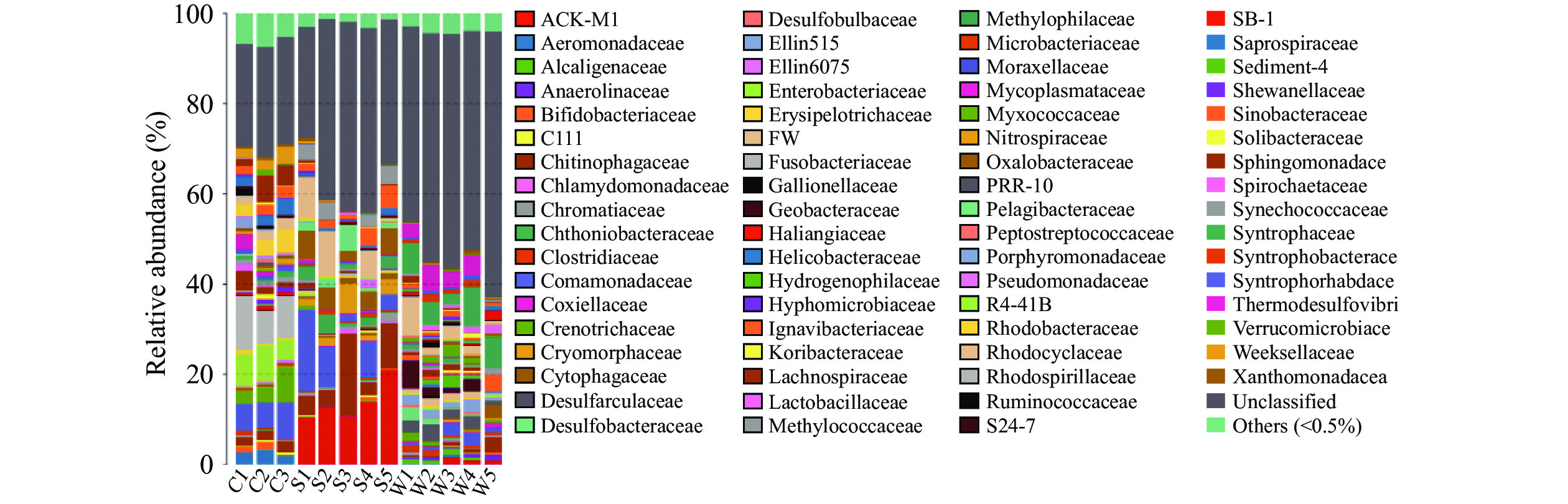
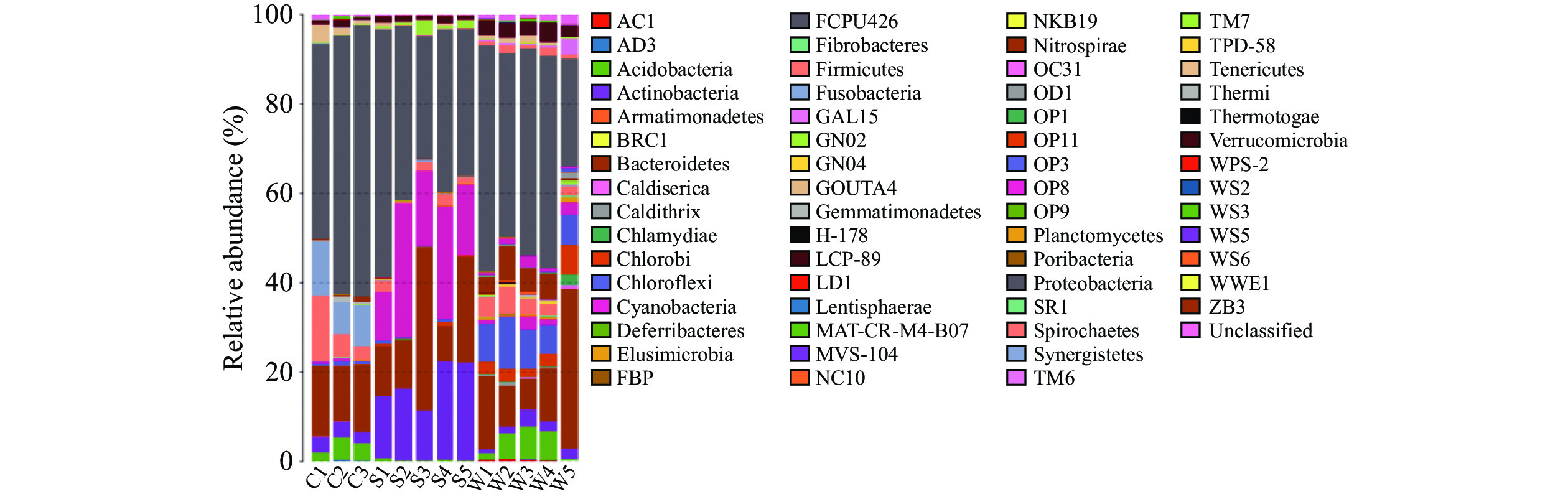
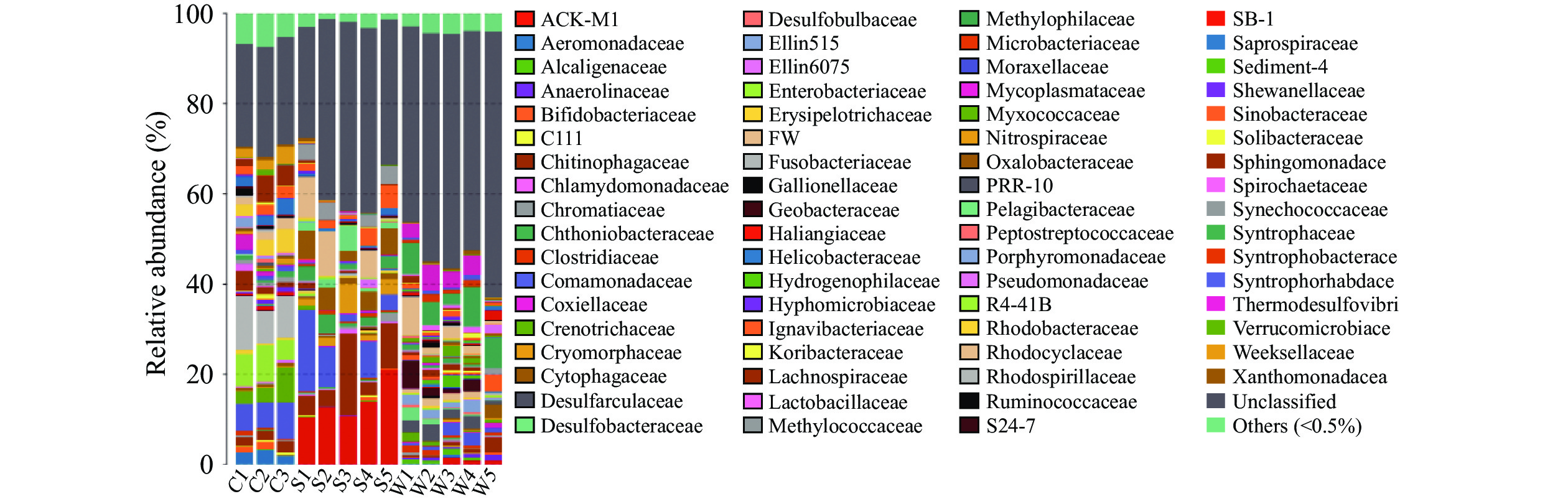
在目、科、属分类水平上,不同样品种类间优势细菌的组成及丰度存在明显差异。在虾肉、土壤及水体中共同的优势目(按>5%记)为伯克氏菌目(Burkholderiales),此外,优势目、科、属在不同样品中各异。在虾肉中梭杆菌目(Fusobacteriales)、甲基球菌目(Methylococcales)、肠杆菌目(Enterobacteriales)显著(P<0.05)高于土壤和水体;在土壤中,优势目则为放线菌目(Actinomycetales和Saprospirales);水体中拟杆菌目(Bacteroidales)、硝化螺旋菌目(Nitrospirales)和红环菌目(Rhodocyclales)显著高于虾肉和土壤(图3)。在科和属分类水平,不同样品种类间优势细菌的组成及丰度差异明显,且无共同优势科和属。从科水平看,小龙虾中主要微生物菌落为梭杆菌科(Fusobacteriaceae)、丛毛单胞菌科(Comamonadaceae)及肠杆菌科(Enterobacteriaceae),土壤中主要微生物菌落为ACK-M1和Chitinophagaceae;水体中主要微生物菌落为互营菌科(Syntrophaceae)、热脱硫弧菌科(Thermodesulfovibrionaceae)以及红环菌科(Rhodocyclaceae)(图4)。在属水平,小龙虾组中的主要优势菌株为鲸杆菌属(Cetobacterium)、泉发菌属(Crenothrix)、邻单胞菌属(Plesiomonas);水体组中的主要优势菌群为4-29、地杆菌属(Geobacter)、HB118;土壤组中的主要优势菌群为聚球藻属(Synechococcus)、C39、Limnohabitans、多核杆菌属(Polynucleobacter)、噬氢菌属(Hydrogenophaga)(图5)。
![]() 图 3 基于目水平的微生物组成分析柱状图Figure 3. Microbial composition analysis histogram based on order level
图 3 基于目水平的微生物组成分析柱状图Figure 3. Microbial composition analysis histogram based on order level![]() 图 4 基于科水平的微生物组成分析柱状图Figure 4. Microbial composition analysis histogram based on family level
图 4 基于科水平的微生物组成分析柱状图Figure 4. Microbial composition analysis histogram based on family level![]() 图 5 基于属水平的微生物组成分析柱状图Figure 5. Microbial composition analysis histogram based on genus level
图 5 基于属水平的微生物组成分析柱状图Figure 5. Microbial composition analysis histogram based on genus level2.4 小龙虾、土壤和水体中具有统计学差异的菌群
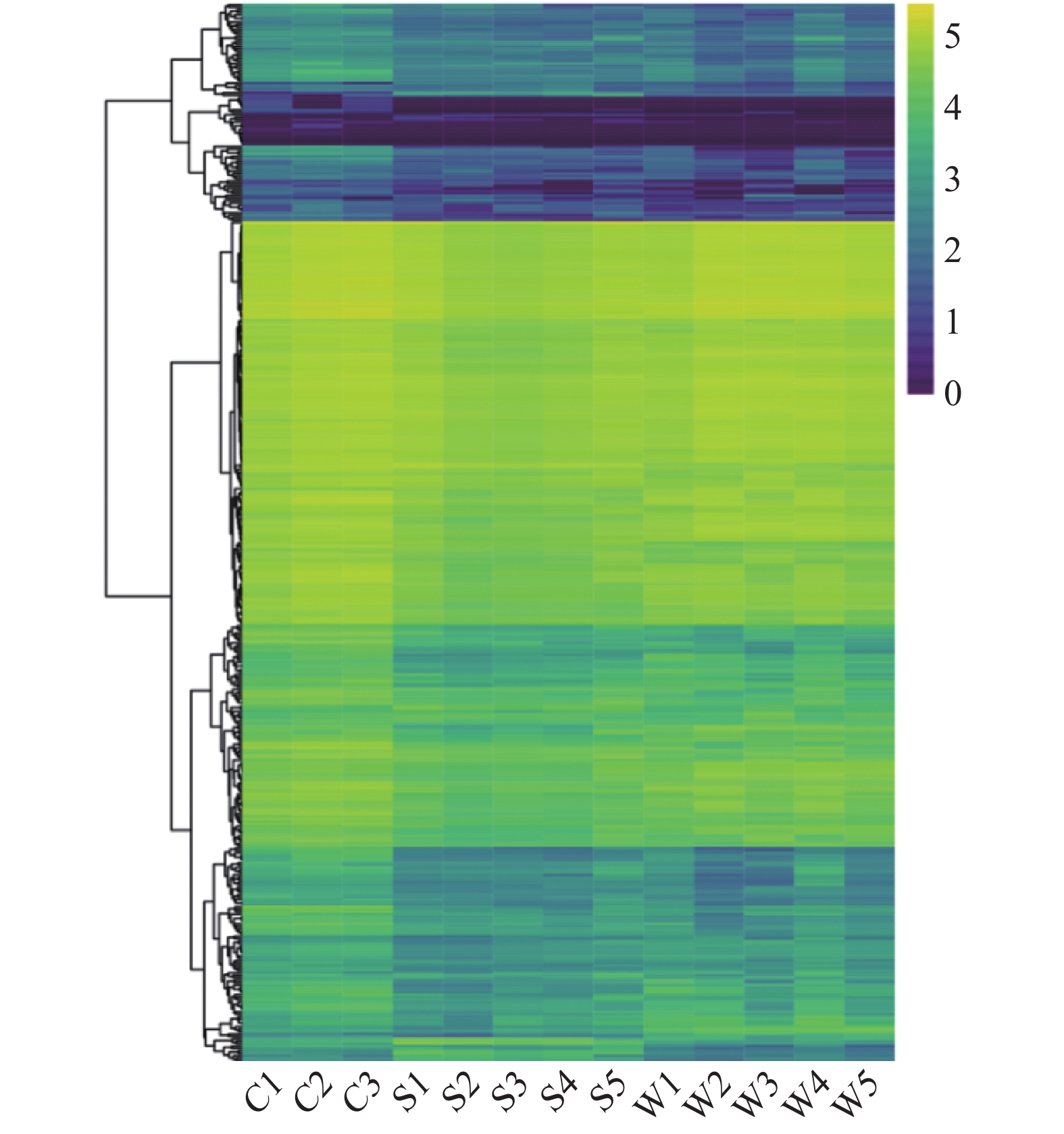
除计算α多样性外,比较微生物群落的一个主要目标是在每个样本中找到特定细菌群。LDAEffectSize分析(Lefse),是一种用于发现和解释高维度数据生物标识(基因、通路和分类单元等)的分析工具[21],能够进行两个或多个分组的比较,强调统计意义和生物相关性,还可以分析组间菌群差异,发现各组间差异的微生物种类(图6A和6B)。使用Lefse软件在每个分类水平上分析细菌群落数据,LDA值为2,共区分出508个细菌群,为清晰起见,分支图谱显示LDA值大于2的分类群,图6A表示三组样品间具有显著性差异的菌群,图6B表示不同差异物种间的进化分支情况,综合分析两组图来反映小龙虾及其生长环境细菌群落差异物种的空间分布特征。在所有样品中,共发现89个差异物种(图6A),小龙虾中共富集了31个类群,其中酸杆菌门、拟杆菌门、绿弯菌门、厚壁菌门、梭杆菌属等相对丰度明显较高;而土壤中共富集了28个类群:酸杆菌门、拟杆菌门、绿菌弯门、厚壁菌门、梭杆菌属、变形菌门、软壁菌门、疣微菌门等;水体中共富集29个类群,包括酸杆菌门、拟杆菌属、绿菌弯门、厚壁菌门、梭杆菌属、变形菌门、软壁菌门、疣微菌门等。
![]() 图 6 Lefse分析显著不同的丰富分类单元注:A:LDA得分分析;B:样品间不同微生物群落的分类图(C:小龙虾,S:土壤,W:水体)。Figure 6. Lefse analyses of the significantly different abundant taxa
图 6 Lefse分析显著不同的丰富分类单元注:A:LDA得分分析;B:样品间不同微生物群落的分类图(C:小龙虾,S:土壤,W:水体)。Figure 6. Lefse analyses of the significantly different abundant taxa2.5 功能预测分析
基于PICRUSt2分析平台的16S功能预测将获得的OTU丰度表结合基因库对微生物的代谢通路进行功能预测(图7)。在所有样品中共检测到433条微生物代谢通路,小龙虾中细菌群落中代谢通路丰度较高的途径主要有脂质代谢、脂肪酸合成、氨基酸合成、有氧呼吸途径、二酰基甘油生物合成、顺式戊酸酯生物合成、丙酮酸发酵途径等;在土壤细菌中代谢通路丰度较高的途径主要有氨基酸合成、有氧呼吸途径、丙酮酸发酵途径、脂肪酸合成、戊糖磷酸途径、TCA循环途径、核苷酸合成途径;在水体细菌中代谢通路丰度较高的途径主要有氨基酸合成途径、糖酵解途径、脂肪酸合成、脂质合成、核苷酸合成途径等。
![]() 图 7 PICRUSt2预测微生物代谢通路丰度图注:C:小龙虾;S:土壤;W:水体。代谢通路丰度越高,颜色越偏向浅色。Figure 7. PICRUSt2 predicted abundance of microbial metabolic pathways
图 7 PICRUSt2预测微生物代谢通路丰度图注:C:小龙虾;S:土壤;W:水体。代谢通路丰度越高,颜色越偏向浅色。Figure 7. PICRUSt2 predicted abundance of microbial metabolic pathways3. 讨论
养殖环境微生物及其菌落结构在水生生物生长过程中具有重要作用,小龙虾体内的微生物与其营养代谢及免疫防御有密切联系,在维持小龙虾生长过程中内部环境稳态发挥作用[22]。对虾肠道的微生物生态被证实与外部水环境密切相关[23]。夏海峰等[24]在研究刺参肠道与养殖池塘底泥微生物关系时发现,随着季节的变化,二者间细菌种群变化趋势相同,说明其菌系具有内在联系。本研究结果显示,小龙虾尾肉、土壤和水体中门、纲、目、科、属在三者间均能被检测,表明小龙虾的养殖环境与小龙虾虾肉间微生物联系密切。在门、纲、目三个分类水平上的分布发现土壤与虾肉间共有微生物较多,而水体分别与土壤及虾肉间共有微生物较少,在科和属分类水平没有检测到共有优势菌群,说明在本研究中,土壤与虾肉中微生物菌落结构在门、纲、目分类水平上密切相关,而在科和属水平无相关性。
分析微生物群落多样性角度可知,小龙虾生长环境中土壤细菌丰富度和多样性最高,而水体中的多样性指数与小龙虾及土壤相比明显较低,这与凡纳滨对虾[25]、青蟹[26]等水生动物的相关研究结果一致。进一步说明小龙虾中的微生物与生长水体及土壤关系密切,其中小龙虾菌群的组成可能较多来源于土壤,符合孙振丽等[27]对南美白对虾的推论,即池塘底泥对南美白对虾肠道菌群的影响大于水体。
在运输储藏过程中,由于生物体内微生物作用,水产品品质会发生不同程度劣变 [28-29]。在微生物中导致产品变质的微生物较少,其中对食品具有高度特异性的腐败细菌为特异性腐败菌。研究表明,海产品中主要优势腐败菌为希瓦氏菌属(Shewanella)和假单胞菌属(Pseudomonas),而淡水产品中主要优势腐败菌为假单胞菌属(Pseudomonas)[30]和芽孢杆菌(Bacillus spp)[31]。江杨阳等[32]利用高通量测序法确定淡水小龙虾冷藏过程中的优势腐败菌为希瓦氏菌(Shewanella)。谢丽丹等[33]发现引起南美白对虾腐败变质的主要细菌是希瓦氏菌(Shewanella hafniensis)和约氏不动杆菌(Acinetobacter johnsonii)。在本研究中,不动杆菌属(Acinetobacter)是小龙虾的优势腐败菌,与先前研究的优势腐败菌略有不同的原因可能是生长环境、贮藏时间及温度的差别导致。不动杆菌被证明在室温下具有较高的脂解活性,并产生硫化氢导致虾肉变质[34]。在水体及土壤中检测到的优势腐败菌为假单胞菌属(Pseudomonas),是水产品储藏过程中产生异味的主要原因[35]。但两种腐败菌种在三组样品中的相对丰度均较小,因此水体及土壤中的微生物会可能会在低温贮藏过程中进行繁殖,影响小龙虾贮藏品质。
PICRUSt(Phylogenetic Investigation of Communities by Reconstruction of Unobserved States)是基于16 sRNA测序数据,通过标记基因数据和参考基因组数据库预测元基因组的功能[36]。在本研究中,基于KEGG同源基因(KO)对细菌功能进行具体注释,结果表明虾肉、土壤和水体共有的细菌优势功能类群为有氧呼吸(aerobic respiration I)、脂肪酸合成(fatty acid elongation)、氨基酸生物合成(amino acid synthesis)和核糖核苷酸代谢途径(nucleotide sugar metabolism),这可能与拟杆菌门分类下的细菌有关。杨盼等[37]发现苜蓿根际土壤中代谢为细菌菌群的主要功能,其中氨基酸代谢丰度较高;凡纳滨对虾水体中微生物功能研究结果显示水体中微生物在细胞新陈代谢及遗传信息处理方面功能基因丰度较高[38]。
4. 结论
本研究采用高通量测序的方法研究了小龙虾及其养殖环境水体和土壤间的微生物菌落分布发现,小龙虾菌群与其生长环境中的土壤和水体中微生物存在密切联系,其中小龙虾和土壤中菌落结构与水体相比较为丰富,三类样品拥有相对固定的优势菌群及共有菌群,包括变形菌门、拟杆菌门等,但同样存在差异物种,此结果为理解三者之间的关联奠定了基础。对小龙虾、土壤和水体中的微生物菌群进行功能注释表明三者间的代谢功能存在高度相似性,证实了小龙虾菌群与其生长环境之间的相关性,其中小龙虾菌落组成可能较多来源于土壤,而水体所含微生物对其影响较小,此研究结果对小龙虾的运输贮藏过程及健康养殖产业具有积极意义。但本研究尚未探讨养殖水体中层水样及下层水样对小龙虾菌落组成的影响,同时对三者之间菌落的关联性未进行深入探讨。
-
![]()
图 1 基于门水平的微生物组成分析柱状图
Figure 1. Histogram of microbial composition analysis based on phylum level
![]()
图 2 基于纲水平的微生物组成分析柱状图
Figure 2. Microbial composition analysis histogram based on class level
![]()
图 3 基于目水平的微生物组成分析柱状图
Figure 3. Microbial composition analysis histogram based on order level
![]()
图 4 基于科水平的微生物组成分析柱状图
Figure 4. Microbial composition analysis histogram based on family level
![]()
图 5 基于属水平的微生物组成分析柱状图
Figure 5. Microbial composition analysis histogram based on genus level
![]()
图 6 Lefse分析显著不同的丰富分类单元
注:A:LDA得分分析;B:样品间不同微生物群落的分类图(C:小龙虾,S:土壤,W:水体)。
Figure 6. Lefse analyses of the significantly different abundant taxa
![]()
图 7 PICRUSt2预测微生物代谢通路丰度图
注:C:小龙虾;S:土壤;W:水体。代谢通路丰度越高,颜色越偏向浅色。
Figure 7. PICRUSt2 predicted abundance of microbial metabolic pathways
表 1 测序结果及质量分析
Table 1 Sequencing results and quality analysis
样品名称 有序数列数量 OTU数量 C1 77858 2619 C2 94552 2548 C3 95353 2635 S1 74143 3497 S2 52230 3734 S3 47921 3737 S4 56102 3278 S5 71336 3274 W1 61734 2596 W2 87664 1789 W3 90037 1871 W4 86817 2648 W5 76027 1813 注:C:小龙虾;S:土壤;W:水体;表2同。  下载: 导出CSV
下载: 导出CSV
表 2 各组样品Alpha多样性统计结果
Table 2 Alpha diversity statistical results for each group
样品名称 Sobs Chao ACE Shannon Simpson C1 2619 3046.880 3036.437 5.565 0.024 C2 2548 2952.909 2892.741 5.760 0.017 C3 2635 3109.000 3029.552 5.580 0.017 S1 3497 4435.584 4532.497 6.008 0.011 S2 3734 4600.083 4746.686 6.721 0.004 S3 3737 4608.254 4732.640 6.767 0.003 S4 3278 3813.233 3918.379 6.580 0.005 S5 3274 4124.446 4136.989 6.016 0.011 W1 2596 3552.264 3625.095 5.206 0.022 W2 1789 2596.072 3126.695 4.364 0.043 W3 1871 2739.095 3352.675 4.748 0.022 W4 2648 3922.967 4820.739 4.712 0.039 W5 1813 2666.415 2788.272 4.601 0.026
下载: 导出CSV
-
[1] 2020中国小龙虾产业发展报告全文发布[J]. 水产科技情报, 2020, 47(4): 229. [2] 叶建勇, 唐金玉, 丁辰龙, 等. 基于高通量测序的克氏原螯虾肠道及其养殖环境菌群结构分析[J]. 青岛农业大学学报(自然科学版),2020,37(2):129−134. [3] Moriarty D J W. The role of microorganisms in aquaculture ponds[J]. Aquaculture,1997,151(1-4):333−349. doi: 10.1016/S0044-8486(96)01487-1
[4] Sun Y, Han W, Liu J, et al. Bacterial community compositions of crab intestine, surrounding water, and sediment in two differentfeeding modes of Eriocheir sinensis[J]. Aquaculture Reports,2020:16.
[5] 秦伟, 周鑫, 周文全, 等. 精养克氏原螯虾池塘底泥微生物群落特征分析[J]. 南方农业学报,2015,46(12):2209−2216. doi: 10.3969/j:issn.2095-1191.2015.12.2209 [6] 王春忠, 林国荣, 严涛, 等. 长毛对虾海水养殖环境以及虾肠道微生物群落结构研究[J]. 水产学报,2014,38(5):706−712. [7] 倪治明. 浙北地区餐饮业小龙虾重点危害因子调查及风险评估[D]. 杭州: 浙江大学, 2013. [8] 李兵兵, 刘纯成, 侯海燕, 等. 淮安地区小龙虾及其外环境中致病菌分布规律和耐药性分析[J]. 食品安全质量检测学报,2016(7):3530−3534. [9] Caporaso J G, Paszkiewicz K, Field D, et al. The Western English Channel contains a persistent microbial seed bank[J]. The ISME Journal,2012,6(2):1089−1093.
[10] 张赫宇, 杨波, 罗瑞明, 等. 高通量测序分析冷鲜滩羊肉储藏过程中的细菌群落多样性[J]. 食品工业科技,2016,37(13):177−182. [11] Jerez C A. The use of genomics, proteomics and other OMICS technologies for the global understanding of biominingmicroorganisms[J]. Hydrometallurgy,2008,94(1-4):162−169. doi: 10.1016/j.hydromet.2008.05.032
[12] Pajarillo E A B, Chae J P, Balolong M P, et al. Characterization of the fecal microbial communities of duroc pigs using 16S rRNA gene pyrosequencing[J]. Asian-Australasian Journal of Animal Sciences,2015,28(4):584−591. doi: 10.5713/ajas.14.0651
[13] 袁钰, 李静, 林少华, 等. 基于16S rDNA高通量测序技术分析北京豆汁儿微生物多样性和功能预测的研究[J]. 食品工业科技,2020,41(2):95−100. [14] 黄锦. 不同施肥模式下稻-克氏原螯虾养殖田块水体、土壤和肠道微生物的研究[D]. 上海: 上海海洋大学, 2019. [15] 汤纯, 诸永志, 吴海虹, 等. 基于传统培养和宏基因组测序分析泗洪小龙虾不同部位的菌群多样性[J]. 肉类研究,2019,33(10):44−50. [16] 伍一宁. CO2浓度升高对洪河自然保护区中小型土壤动物及微生物群落生态影响研究[D]. 哈尔滨: 东北林业大学, 2019. [17] Zhang M, Liu W, Nie X, et al. Molecular analysis of bacterial communities in biofilms of a drinking water clearwell[J]. Microbes & Environments,2012,27(4):443−448.
[18] Edgar R C, Haas B J, Clemente J C, et al. UCHIME improves sensitivity and speed of chimera detection[J]. Bioinformatics,2011,27(16):2194−2200. doi: 10.1093/bioinformatics/btr381
[19] Qiong W, M G G, M T J, et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J]. Applied and Environmental Microbiology,2007,73(16):5261−5267. doi: 10.1128/AEM.00062-07
[20] Liu Z, Li J, Huang T, et al. Comparison of the bacterial communities in home-made Nanfengyancai with and without salt[J]. Food Research International,2019,125:108509.1−108509.12.
[21] Langille M, Zaneveld J, Caporaso J G, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences[J]. Nature Biotechnology,2013,31(9):814−821. doi: 10.1038/nbt.2676
[22] 朱旭. 秸秆还田与投食对稻虾共作系统土壤硝化作用及微生物的影响[D]. 武汉: 华中农业大学, 2020. [23] 李可, 郑天凌, 田蕴, 等. 南美白对虾肠道微生物群落的分子分析[J]. 微生物学报,2007(4):649−653. doi: 10.3321/j.issn:0001-6209.2007.04.016 [24] 夏海峰, 杜宗军, 陈冠军. 刺参肠道及养殖池塘底泥微生物多样性的比较研究[J]. 海洋湖沼通报,2015(4):105−110. [25] 杜世聪, 黄雷, 杨坤杰, 等. 凡纳滨对虾健康状态分化前后养殖水体浮游细菌群落的比较[J]. 生态学杂志,2019,38(8):2456−2465. [26] 王贤丰, 赵艳飞, 宋志飞, 等. 应用高通量测序技术分析拟穴青蟹肠道及其养殖环境菌群结构[J]. 中国水产科学,2017,24(6):1245−1253. [27] 孙振丽, 宣引明, 张皓, 等. 南美白对虾养殖环境及其肠道细菌多样性分析[J]. 中国水产科学,2016,23(3):594−605. [28] 刘晓畅, 罗永康. 水产品贮运过程品质预测技术研究进展[J]. 中国渔业质量与标准,2016,6(2):1−6. [29] 杨宪时, 许钟, 肖琳琳. 水产食品特定腐败菌与货架期的预测和延长[J]. 水产学报,2004(1):106−111. [30] 张爱萍, 刘翔, 李永才, 等. 水产品储运过程中的防腐保鲜[J]. 包装与食品机械,2009,27(5):116−118. doi: 10.3969/j.issn.1005-1295.2009.05.032 [31] 邓灵, 赵康, 夏开, 等. 小龙虾(Procambarus clarkii)加工前后优势腐败菌的分离与鉴定[J]. 食品工业科技,2020,41(18):100−104. [32] 江杨阳, 杨水兵, 余海霞, 等. 基于培养基法和高通量测序法分析冷藏小龙虾优势腐败菌[J]. 食品科学,2019,40(16):130−136. doi: 10.7506/spkx1002-6630-20180718-229 [33] 谢丽丹, 李蕾蕾, 王素英, 等. 低温贮藏南美白对虾特定腐败菌的分离鉴定及腐败能力分析[J]. 食品与发酵工业,2016,42(3):67−72. [34] Sahna D, Martin X K A, Tuni D S, et al. Identification of potential spoilage bacteria in farmed shrimp (Litopenaeus vannamei): Application of relative rate of spoilage models in shelf life-prediction[J]. LWT- Food Science and Technology,2018,97:295−301. doi: 10.1016/j.lwt.2018.07.006
[35] Powell S M, Tamplin M L. Microbial communities on Australian modified atmosphere packaged Atlantic salmon[J]. Food Microbiology,2012,30(1):226−232. doi: 10.1016/j.fm.2011.10.002
[36] Chen Y, Tian W, Shao Y, et al. Miscanthus cultivation shapes rhizosphere microbial community structure and function as assessed by Illumina MiSeq sequencing combined with PICRUSt and FUNGUIld analyses[J]. Archives of Microbiology,2020,202(5):1157−1171. doi: 10.1007/s00203-020-01830-1
[37] 杨盼, 翟亚萍, 赵祥, 等. 丛枝菌根真菌和根瘤菌互作对苜蓿根际土壤细菌群落结构的影响及PICRUSt功能预测分析[J]. 微生物学通报,2020,47(11):3868−3879. [38] 张哲, 杨章武, 葛辉, 等. 凡纳滨对虾育苗水体中三种生物絮团的菌群多样性及Tax4Fun基因功能预测分析[J]. 水生生物学报,2019,43(4):786−796. doi: 10.7541/2019.093
 下载:
下载:







计量
- 文章访问数: 235
- HTML全文浏览量: 89
- PDF下载量: 23
